Supercomputadoras para el diseño de fármacos
Diego Masone. CONICET - Facultad de Ciencias Exactas y Naturales, Universidad Nacional de Cuyo.
La mayor parte de los procesos biológicos que tienen lugar en los seres vivos involucran interacciones entre proteínas, estas agrupaciones organizadas de aminoácidos que cumplen funciones vitales de la célula. Es así que la correcta identificación y caracterización de las interacciones entre proteínas y el conocimiento de la estructura y las propiedades de los complejos que forman, resulta crucial a la hora de comprender cómo funcionan las células. Este tipo de problema es abordado desde hace décadas utilizando modelos computacionales para el desarrollo de métodos de diseño de nuevos fármacos. Aunque no podía anticiparse en 1938, el problema se parece al descripto por Albert Einstein y Leopold Infeld en el inolvidable libro “La evolución de la física”. Dice así:
“...los conceptos físicos son creaciones libres del espíritu humano y no están, por más que lo parezca, determinados unívocamente por el mundo exterior. En nuestro empeño de concebir la realidad, nos parecemos a alguien que tratara de descubrir el mecanismo invisible de un reloj, del cual ve el movimiento de las agujas, oye el tic-tac, pero no le es posible abrir la caja que lo contiene. Si se trata de una persona ingeniosa e inteligente, podrá imaginar un mecanismo que sea capaz de producir todos los efectos observados; pero nunca estará segura de si su imagen es la única que los pueda explicar. Jamás podrá compararla con el mecanismo real, y no puede concebir, siquiera, el significado de una tal comparación. Como él, el hombre de ciencia creerá ciertamente que, al aumentar su conocimiento, su imagen de la realidad se hará más simple y explicará mayor número de impresiones sensoriales. Puede creer en la existencia de un límite ideal del saber, al que tiende el entendimiento humano, y llamar a este límite la verdad objetiva.”
En la segunda mitad del siglo XIX el límite de resolución de un microscopio óptico había sido definido matemáticamente por Ernst Karl Abbe y se había alcanzado el punto en que ya no era posible ir más allá de las observaciones de medio micrómetro (0,0000005 m). Sin embargo el desarrollo del tubo de rayos catódicos impulsó el gran salto en microscopía: usando electrones en lugar de luz visible Gustav Ludwig Hertz demostró teóricamente cómo un pequeño solenoide podía hacer converger haces de electrones de forma análoga a como una lente convergía rayos de luz visible.
Aunque originalmente concebido por Leó Szilárd en 1933, los ingenieros alemanes Ernst Ruska y Maximillion Knoll construyeron el primer microscopio electrónico que tenía una resolución de 50 nanómetros (0,000000050 m). Lamentablemente el estallido de la Segunda Guerra Mundial retrasó los avances y sólo después de 20 años de terminada la guerra los microscopios electrónicos fueron capaces de alcanzar 1 nanómetro de resolución (0,000000001 m). Estos fueron los primeros pasos hacia las técnicas de cristalografía por rayos X con las que hoy se estudian las proteínas en gran detalle.
En un esfuerzo por almacenar, organizar y compartir información cristalográfica la base de datos de proteínas de libre acceso en internet (véase www.pdb.org) fue creada en los Laboratorios Nacionales de Brookhaven (BLN) en 1971. En sus comienzos contenía sólo 13 estructuras, hoy cuenta con más de 100.000. Esto significa que la humanidad dispone de la información estructural de esta cantidad de proteínas a sólo un click de distancia, muchas de ellas responsables directa o indirectamente de las más horribles patologías y enfermedades. Es sin duda una situación sin precedentes para la investigación biomédica.
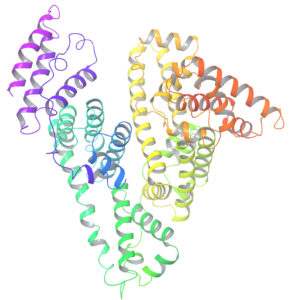 Es entonces muy fácil para nosotros descargar de internet una proteína de nuestro agrado y “ver” cómo luce su disposición tridimensional en una computadora de escritorio, como muestra la figura 1. Pero podemos hacer mucho más, y de eso se encarga la biomedicina computacional. Podemos estudiar cómo interacciona nuestra proteína con otras proteínas o cómo lo hace con pequeñas moléculas. Podemos diseñar nuestras propias moléculas en la computadora y simular su interacción con nuestra proteína para por ejemplo, inhibir cierta función relacionada con alguna enfermedad. Es decir, podemos diseñar fármacos virtuales que luego sean probados experimentalmente hasta llegar a convertirse en fármacos reales.
Es entonces muy fácil para nosotros descargar de internet una proteína de nuestro agrado y “ver” cómo luce su disposición tridimensional en una computadora de escritorio, como muestra la figura 1. Pero podemos hacer mucho más, y de eso se encarga la biomedicina computacional. Podemos estudiar cómo interacciona nuestra proteína con otras proteínas o cómo lo hace con pequeñas moléculas. Podemos diseñar nuestras propias moléculas en la computadora y simular su interacción con nuestra proteína para por ejemplo, inhibir cierta función relacionada con alguna enfermedad. Es decir, podemos diseñar fármacos virtuales que luego sean probados experimentalmente hasta llegar a convertirse en fármacos reales.
Desde luego, detrás de este proceso computacional están las leyes físicas y químicas que gobiernan los átomos que componen los aminoácidos y que su vez forman las proteínas. La dinámica de moléculas es en términos computacionales un experimento virtual donde hemos incluido todo lo que sabemos sobre fuerzas interatómicas. Es el descomunal poder de cálculo de una máquina quien simula el paso del tiempo para que observemos la evolución de nuestro sistema virtual y podamos dilucidar sus mecanismos de funcionamiento, detectar problemas y proponer modelos. Con este fin se han construido centenares de supercomputadoras de uso académico en diversas universidades del mundo (véase www.top500.org).
Siguiendo esta línea de investigación hemos desarrollado junto a nuestros colaboradores del Centro Nacional de Supercomputación en Barcelona (véase www.bsc.es), un método de análisis de complejos entre proteínas. Dado un par de proteínas que sabemos se asocian para formar una nueva estructura y de la cual poseemos información cristalográfica detallada, generamos de forma virtual centenares de posibles complejos según un criterio geométrico. Luego, para decidir cuales de ellos son factibles de existir en la naturaleza, utilizamos un tipo muy especial de interacción intermolecular: los enlaces de hidrógeno. Este tipo de interacción resulta sumamente importante durante el reconocimiento molecular como se ha demostrado experimentalmente mediante cálculos de estructura electrónica.
De esta forma extendiendo el proceso a una buena cantidad de proteínas distintas, verificamos que los cálculos de energías de interacción que incluían una completa optimización de los enlaces de hidrógenos de las proteínas en su totalidad, introducían una significativa mejora en el discernimiento entre los complejos generados geométricamente. Esto significa que luego de generar en una computadora centenares de complejos entre proteínas de una manera más bien caprichosa, somos capaces de distinguir cuales tienen una buena posibilidad de existir realmente y con un poco de suerte, tendremos un número reducido de candidatos.
Este último conjunto de estructuras es hasta donde la biología computacional puede llegar (por ahora) y que a continuación sigue la demoledora etapa experimental. Pero no es difícil imaginar un futuro con diseño de fármacos “a la carta” específicos para cada necesidad. Como “Los cazadores de microbios” de Paul de Kruif, queda en manos de los científicos la búsqueda de la verdad objetiva que nos permita avanzar en el entendimiento de las interacciones entre proteínas.
Contacto: [email protected]
Artículo original: H-bond network optimization in protein–protein complexes: Are all-atom force field scores enough? Diego Masone, Israel Cabeza de Vaca, Carles Pons, Juan Fernandez Recio and Victor Guallar. Proteins: Structure, Function, and Bioinformatics Volume 80, Issue 3, pages 818–824, March 2012. http://onlinelibrary.wiley.com/doi/10.1002/prot.23239/abstract